Automatically Link Large-scale Molecular Dynamics ASE for Simulation¶
The neural network potential function whose training accuracy meets the requirements of first principles can realize large-scale molecular dynamics simulation through the Pmlib interface. The visual interface of this interface is shown in the figure.

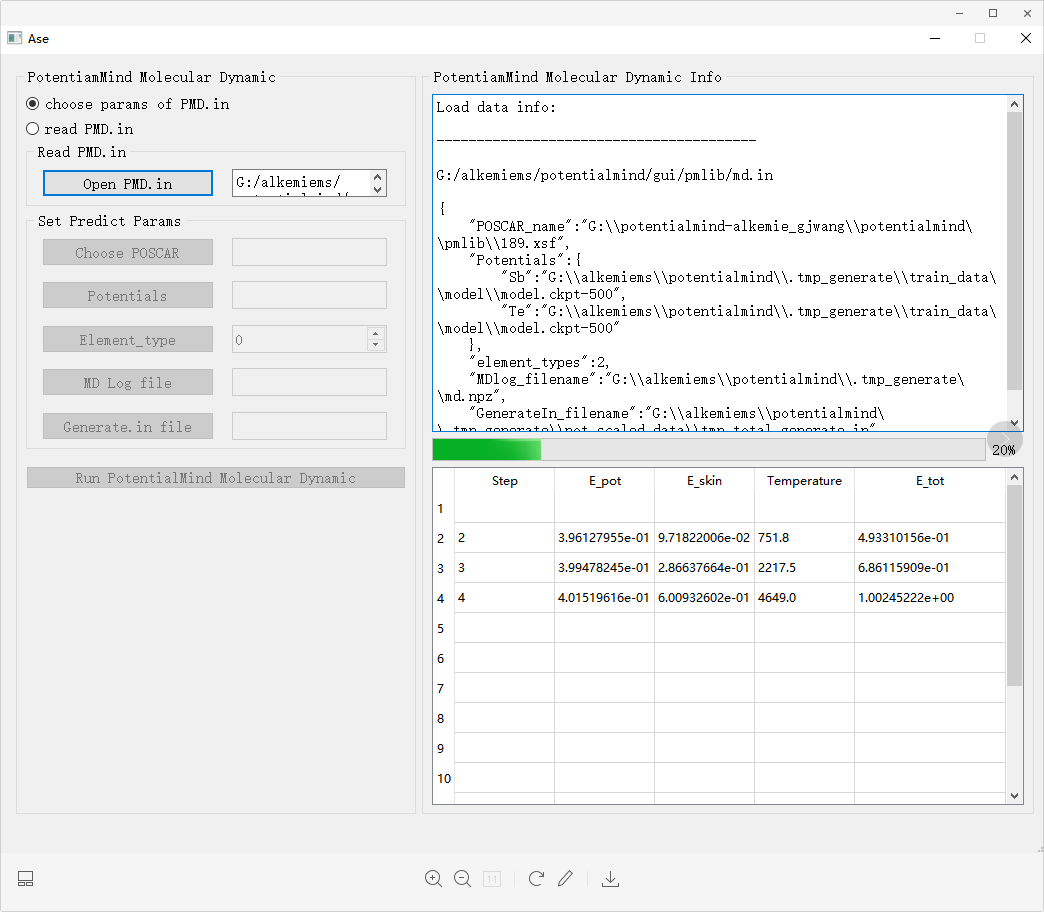
First, the user needs to specify the parameters used in the training process, or directly read the existing large-scale molecular dynamics parameters. To run large-scale molecular dynamics, the initial POSCAR structure must be entered. Second, specify the position of the trained potential function model corresponding to each element in the structure. Then select the MDLog file that saves the key parameters during the potential function training process. Currently, only the read parameter part has been developed, and specific parameters are yet to be developed.