VASP Workflow Control¶
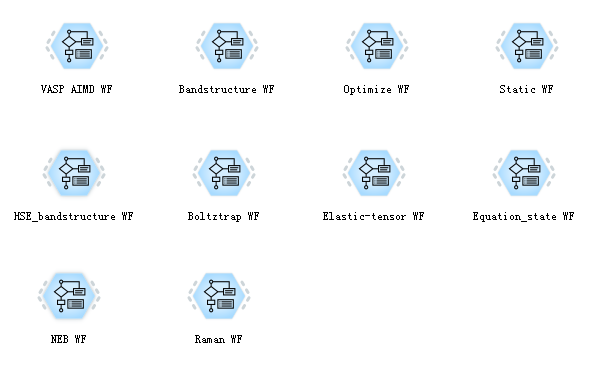
The software supports a total of 10 basic workflows, including structural optimization, energy band structure calculation, density of state calculation, molecular dynamics simulation, HSE energy band structure calculation, and so on.
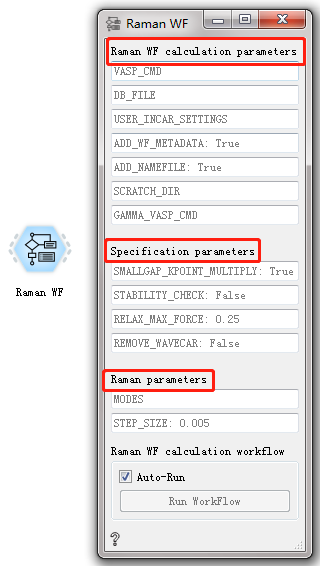
Each workflow has input and output files and corresponding parameter settings. There are generally three groups of parameter configuration: general calculation parameters, specific calculation parameters and workflow parameters. All parameters have common default settings, which is very convenient for novice users. Double-click to open the workflow, and you can see that each workflow contains general calculation parameters and specific calculation parameter settings. The two sets of parameter settings will be introduced in detail below.
General calculation parameters:
VASP_CMD set vasp calculation method
DB-FILE use the database configuration file for calculation
USERINCARSETTINGS customizable INCAR calculation parameters
ADDWFMETADATA: True add structural metadata to the workflow by default
ADD_NAMEFILE: True a file name "FW——<FW.name>" is automatically generated before the start of each calculation task, which is used to save all the running directories for searching
SCRATCH_DIR the save directory of the previously calculated file is the current directory by default
GAMMAVASPCMD set the operation mode of Vasp on the remote computer, or control the storage location of Vasp executable file
Specific calculation parameters:
SMALLGAPKPOINTMULTIPLY: True use a lot of small KPOINTS in the workflow by default
STABILITY_CHECK: False do not check the stability of the structure by default
RELAXMAXFORCE: 0.25 the maximum atomic force accuracy for structural convergence is 0.25
REMOVE_WAVECAR: False WAVECAR is not deleted by default
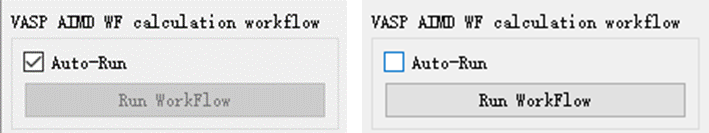
In addition, there is an Auto-Run option at the bottom of each workflow parameter setting window. If you check this option, the workflow can start running automatically; if you don’t check this option, you can start running by clicking Run Workflow.
After introducing the common configuration of each workflow, we will sequentially introduce the purpose of individual controls, input and output files, and its unique workflow parameter settings.
VASP AIMD WF CONTROL¶
Used to calculate molecular dynamics.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the VASP AIMD WF workflow has three sets of parameter configurations: General calculation parameters, specific calculation parameters, AIMD workflow parameters.
For general calculation parameters and specific calculation parameter settings, please refer to the overview of workflow controls. see this!
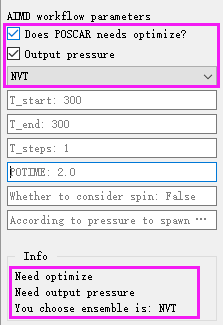
AIMD workflow parameter setting:
Does workflow parameters You can choose whether POSCAR needs structural optimization.
Output presure Whether to output pressure value
NVT / NVE / NPT select system
T_start The default start time is 300
T_end The default end time is 300
T_steps The default time step is 1
POTIME The default is 2
Whether to consider spin: False Spin is not considered by default
According to pressure to spawn MD: False Pressure does not cause molecular motion by default
Info The information bar displays the AIMD workflow parameters after the setting is completed for confirmation
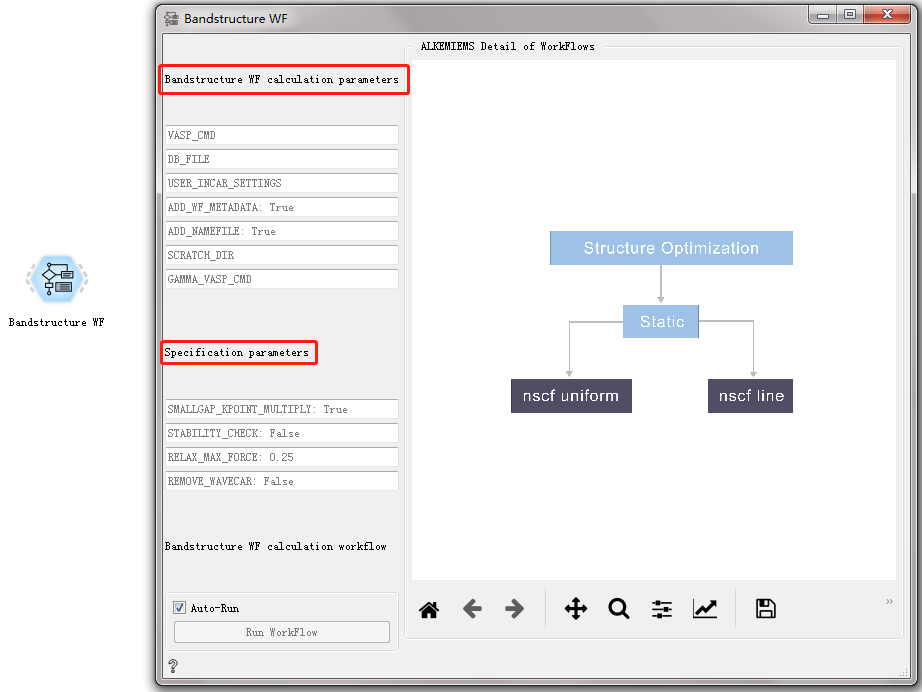
Bandstructure WF CONTROL¶
Used to calculate the energy band structure of the material.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Bandstructure WF workflow only has general calculation parameters and specific calculation parameters. For detailed introduction, please refer to Overview of Workflow Controls. see this!
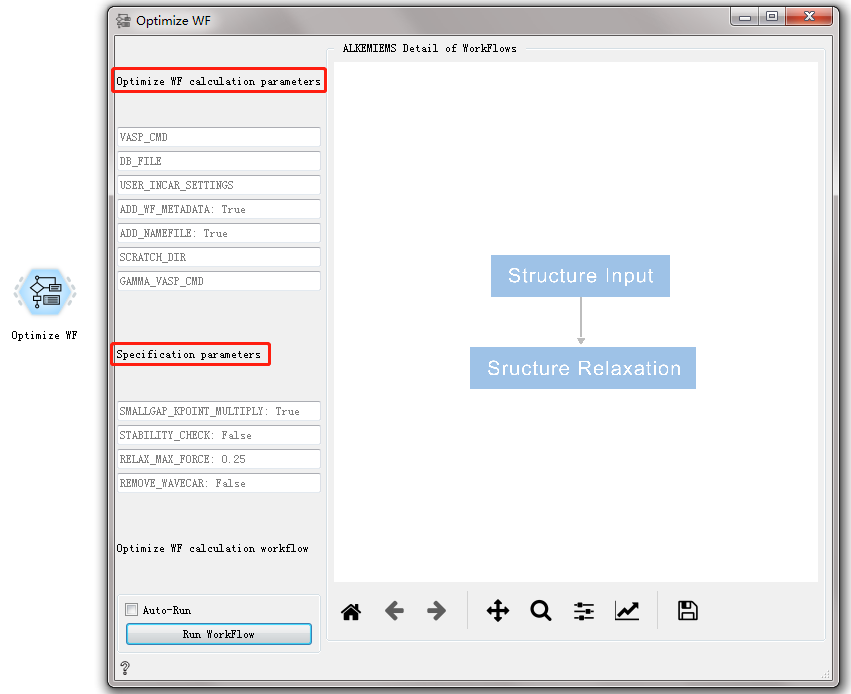
Optimize WF CONTROL¶
Used for structural optimization.
Input |
Output |
||||||
|---|---|---|---|---|---|---|---|
Structure |
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Optimize WF workflow is the same as the Bandstructure WF workflow, with only general calculation parameters and specific calculation parameters. For detailed introduction, please refer to Overview of Workflow Controls. see this!
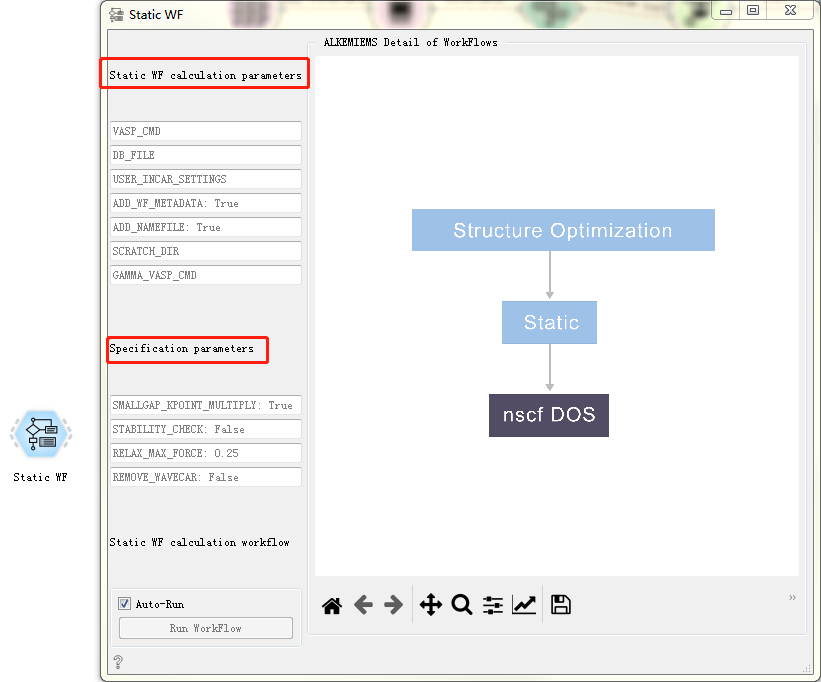
Static WF CONTROL¶
Used for static calculations.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Static WF workflow only has general calculation parameters and specific calculation parameters. For detailed introduction, please refer to Overview of Workflow Controls. see this!
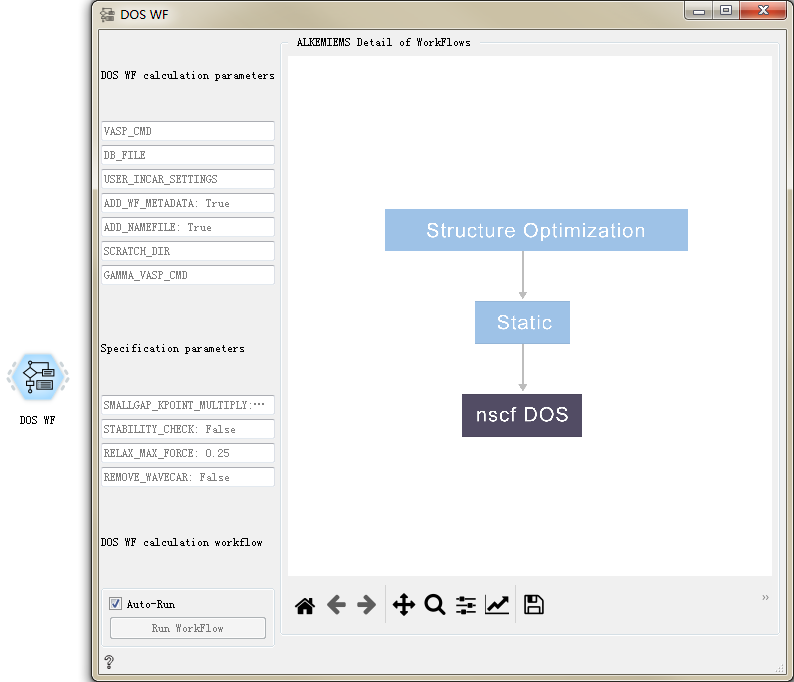
DOS WF CONTROL¶
Used to calculate the electronic density of states of materials.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the DOS WF workflow only has settings for general calculation parameters and specific calculation parameters. For detailed introduction, please refer to Overview of Workflow Controls. see this!
HSE_bandstucture WF CONTROL¶
The energy band structure of the material is calculated by the method of hybrid density functional (HSE).
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the HSE_bandstucture WF workflow only has settings for general calculation parameters and specific calculation parameters. For detailed introduction, please refer to Overview of Workflow Controls. see this!
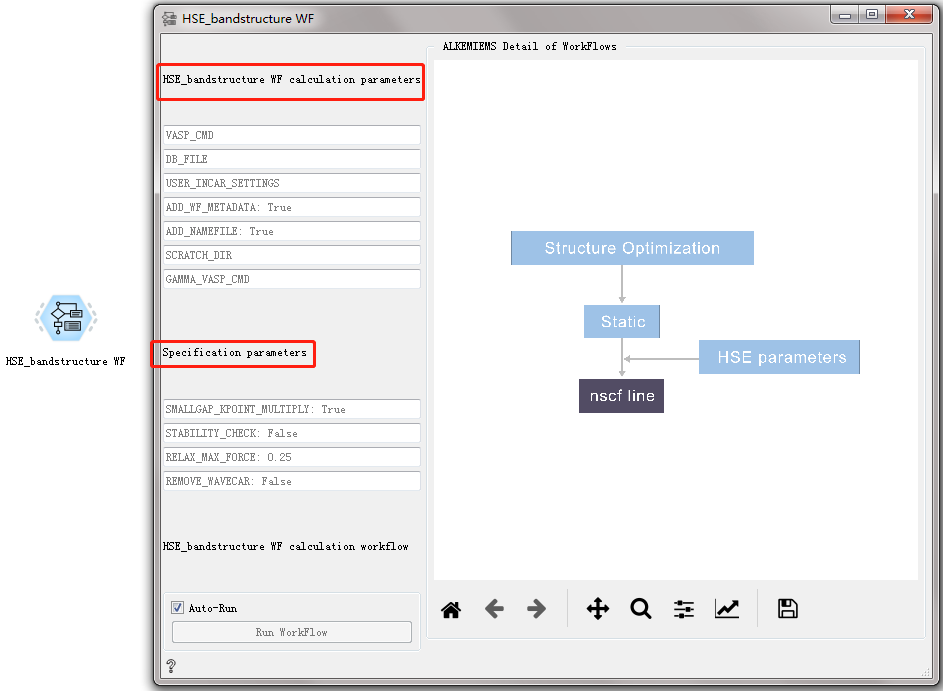
Boltztrap WF CONTROL¶
Used to calculate the Boltzmann transport characteristics of the material.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Boltztrap WF workflow only has settings for general calculation parameters and specific calculation parameters. For detailed introduction, please refer to Overview of Workflow Controls. see this!
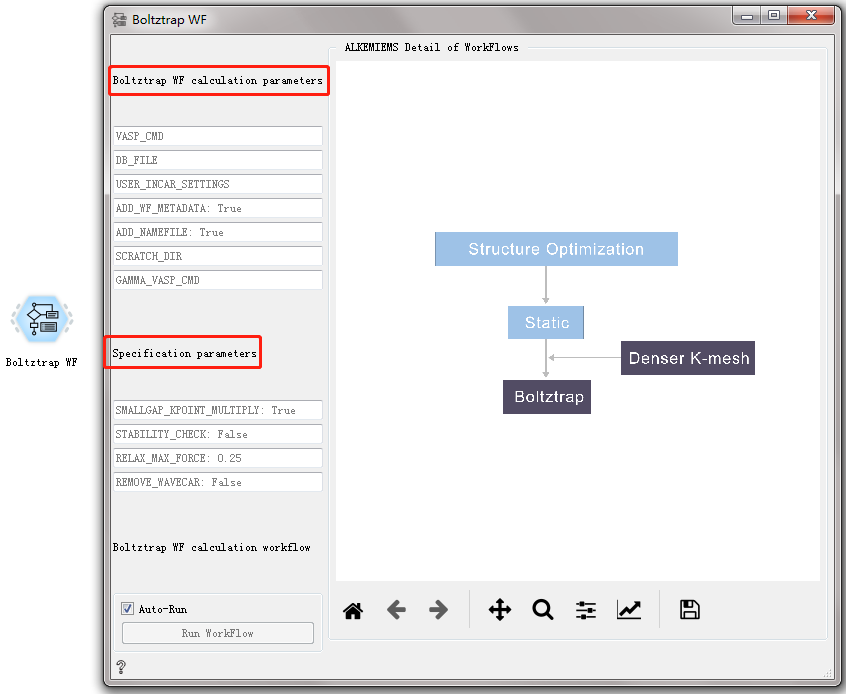
Elastic-tensor WF CONTROL¶
Used to calculate the elastic modulus of the material.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Elastic-tensor WF workflow has three sets of parameter settings: General calculation parameters, specific calculation parameters, and Elastictensor calculation parameters. For general calculation parameters and specific calculation parameters, please refer to the overview of workflow controls. see this!
Elastictensor parameters:
order: 2
kpt: 7000
sym_reduce: False do not change the symmetry of the structure by default
Equation_state WF CONTROL¶
Used to calculate the equation of state of the material.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Equation_state WF workflow has three sets of parameter configurations: General calculation parameters, specific calculation parameters, and Equationstate workflow parameters. Please refer to the general description of workflow controls for general calculation parameters and specific calculation parameter settings. see this!
Equationstate perameters:
EOS set state equation
QHA_TYFE
T_MIN minimum time
T_MAX maximum time
T_STEP set time step
PRESSURE set pressure
POISSON set Poisson distribution
ANHARMONIC_CONTRIBUTION
METADATA Metadata
DEFORMATIONS deformation
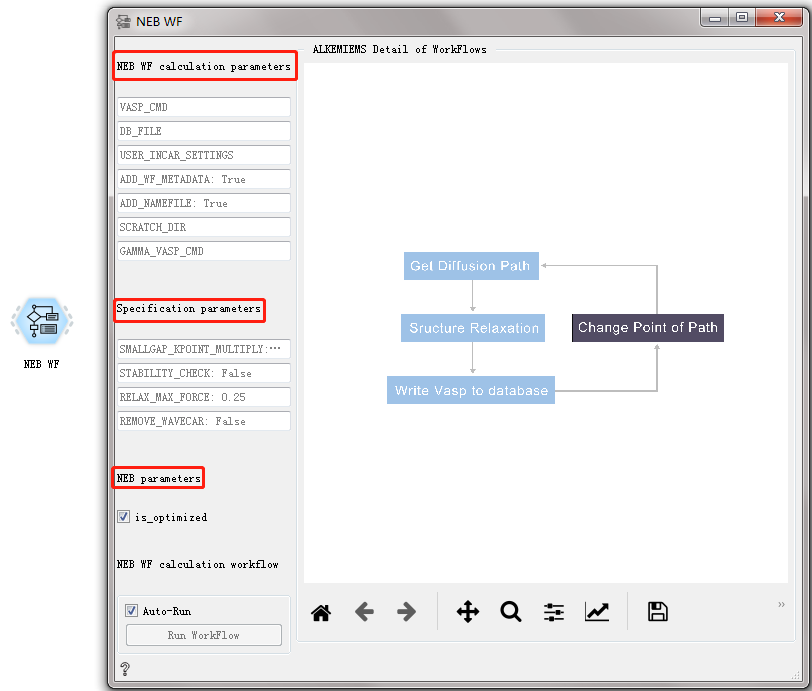
NEB WF CONTROL¶
Used to calculate the transition state of the material.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the NEB WF workflow has three sets of parameter configurations: General calculation parameters, specific calculation parameters, and NEB workflow parameters. For general calculation parameters and specific calculation parameter settings, please refer to the overview of workflow controls. see this!
NEB perameters:
is_optimized You can choose whether the material structure is an optimized structure.
Raman WF CONTROL¶
Used to calculate the Raman spectrum of the material.
Input |
Output |
|||||
|---|---|---|---|---|---|---|
POSCAR file |
INCAR |
POTCAR |
KPOINTS |
Data |
Workflow |
Workflow list |
After double-clicking to open the control, you can see that the Raman WF workflow has three sets of parameter configurations: General calculation parameters, specific calculation parameters, and Raman parameters. Please refer to the general description of workflow controls for general calculation parameters and specific calculation parameter settings. see this! Raman perameters:
MODES set calculation mode
STEP_SIZE: 0.005 the default step size is 0.005